Biomarkers and Causality: A fundamental problem?
(Disclaimer: This article is for informational purposes and does not constitute medical advice. I am not a doctor.)
Biomarkers, e.g. concentrations of various compounds (e.g. glucose, LDL) in the blood, are heavily used in the decision making in modern medicine. For example, those with a high level of blood glucose are prescribed periodic insulin shots and those with a high level of LDL are prescribed daily statins. Sometimes, biomarkers can catch some disease years/decades before any symptoms show up. On the flip side, a misunderstanding of the limitations of biomarkers has led to some of the biggest blunders of modern medicine, causing avoidable misery and/or early death of millions of people. In this article, I deep-dive into 2 cases of these misunderstandings, which in my opinion have been the most damaging to worldwide health. Using these and other examples, I illustrate 3 fundamental problems with modern medicine’s reliance on biomarkers:
- Just because a biomarker is associated with some adverse outcomes AND reducing it by some techniques improves outcomes, doesn’t mean the biomarker causes the problem.
- Even when the biomarker is causal, it may not be the ROOT cause of the problem, in which case, “optimizing” the biomarker may worsen the ROOT cause of the problem in the long run.
- In some cases, there is no good scientific technique to ascertain whether a biomarker is causal.
Citing policy at this blog:
I have been reading the medical literature intensively for over two years now. Very often, I encounter papers, often very influential ones (e.g. those that form the basis of guidelines for doctors around the world), that cite a paper (say X) for a claim (say C) and when I read the paper X, I find that X’s claim is much weaker and sometimes even the exact opposite of C. Avoiding such flaws for the claims made here is an important goal of this blog. So, this blog has the following policy for whenever a paper X is cited/linked for the claim C:
- either the title or abstract of X very unequivocally makes the claim C
- or the part of X that supports C is buried deep inside the paper. In that case, the link at this blog takes you to either the specific table or picture, or a local page at this blog with screenshot(s) of the part(s) of X that make the claim C and a link to the official copy of X. The screenshots highlight the relevant bits, yet provide at least a paragraph of context.
Case1: Glucose in Diabetes
For decades, a high glucose level has been used as the main criteria for diagnosing diabetes, a disease that humans have known about for thousands of years, as evidenced by the following quote from this article
Around the 5th century BC, the famous Indian surgeon Sushruta, in his work Samhita, identified diabetes, by using the term madhumeha (honey-like urine) and pointed out not only the sweet taste of the urine but also its sticky feeling to the touch and its ability to attract the ants (!). Sushruta further mentions that diabetes affects primarily the rich castes and is related to the excessive food consumption as the rice, cereals, and sweet.
Until insulin was discovered, in many parts of the world, diabetes was managed/reversed successfully by reduction of sugar and starchy foods. Here is a snippet from the 1930 book Every Woman’s Doctor Book

There are numerous other such books/accounts prior to the 1950s of successful reversal/management of diabetes using low-starch, high animal protein and/or animal fat diets: e.g. here, an 1895 book which includes many testimonials of successes. Here is a diabetic recipe book from 1917. It is safe to say though that these practices and successes were not widely known.
Later during the last century, LDL phobia took over and starch became a superfood. Also scientists discovered insulin and a way to produce it for external injection. With a perfectly timed and dosed insulin injection, you can keep your blood glucose concentration low even if you eat starchy foods. Soon, insulin injections – rather than low-starch diets– became the de-facto standard for managing diabetes.
There is plenty of evidence that the high glucose level is not the root cause of diabetes. It is likely a result of fat cells being overfilled, resulting in their refusal to take up the excess glucose from the bloodstream. For example, implanting subcutaneous fat in mice (with limited fat cells) seems to cure their diabetes. This also explains why in humans, significant weight loss, even by using entirely opposite strategies (calorie-wise >90% carb (rice and sugar) or <15% carb) reverses diabetes. If diabetes is indeed caused by overfilled fat cells, injecting more insulin, which forces glucose into existing fat cells (instead of adding new fat cells), would only make things worse for diabetics in the long run, a phenomenon widely seen in practice.
Although significant weight loss usually reverses diabetes usually regardless of the method, many of my friends, family, and I find it much easier to lose weight on low-carb high-fat diets because of a much better control on hunger than on high-carb, low fat diets. Many doctors have reported the same in their patients. In contrast, the doctor in the rice and sugar diet trial (>90% carbs) admitted to assaulting his patients into compliance. Perhaps this is the reason diabetics were advised low starch/sugar diets several decades ago. Nevertheless, it seems that some people thrive on plant-based carb-heavy diets and we should be happy about that. The goal of nutrition science to should be to give people as many choices as possible. Unfortunately, the current state seems more geared towards minimizing choices based on anti-animal, anti-plant, anti-carb, anti-fat, anti-xxx dogmas. Food is a great source of pleasure for many people. Don’t believe me? you may change your mind if you do a 3+ day fast.
In type-2 diabetics, high glucose usually follows high insulin levels, at least in the early stages. Without many experiments to determine whether the complications correlated with high glucose were actually caused by high glucose levels, scientists/doctors around the world believed this for decades. There were some experiments trying to distinguish the mechanistic role of high glucose and high insulin, but these seem to be largely ignored by the mainstream guidelines:
Hyperglycemia stimulates coagulation, whereas hyperinsulinemia impairs fibrinolysis in healthy humans.
In this study, by continuous measurement and injection of glucose or insulin, they subjected “healthy” humans to either high glucose levels or high insulin levels (but not both) and observed the changes in various biomarkers in the blood. Those with higher blood glucose but low/normal insulin had signs of increased blood clotting. It is not clear whether blood clots cause damage or blood clots happen in response to damage, or both. Regardless, blood clots can block blood flow in arteries and can cause serious emergencies like heart attacks, strokes.
As this analysis of hearts of 142 dead men revealed, not all ruptures/damages/clots inside arteries are fatal. Our bodies can cover the clot in the outer (adjacent to blood flow) membrane of arteries by a new layer of membrane cells and start healing the clot under the new layer. The human experiment described above suggests that high insulin impairs fibrinolysis, even when glucose is low. Fibrinolysis is the process of dissolving the clot (a fibrin mesh gives structure/support to clots) during/after the repair is complete, so that the normal blood flow can be restored. This process was studied in detail in animals and in-vitro (in lab, not inside a living body) decades ago, e.g. here. Thus, it is likely that high insulin worsens heart disease by impairing the healing of lesions inside arteries. Not just heart disease, high insulin is observationally associated and suspected to be mechanistically causal in a wide range of diseases: joint degeneration, cancer, eye damage, fatty liver disease, kidney disease, Alzhemer’s disease, dementia, osteoporosis. If so, treating type-2 diabetics with insulin is like treating alcoholics with alcohol.
In biology, mechanistic theories, in-vitro (lab) studies, and biomarkers have a long history of being misleading. So, the above arguments should be taken with a grain of salt. However, many of their predictions have been observed in clinical practice and in Randomized Controlled Trials (RCTs). For example, It is well known that in the long run, insulin injections don’t reverse or even stop the progress of the disease. Insulin dosage gradually goes up and up and the patient eventually dies with a diabetes complication. No wonder, diabetes is considered by the medical mainstream as a progressive disease.
In the ACCORD study, which was an RCT, one group called the intensive group (with randomly selected patients) used more agressive therapies (mainly higher insulin) to achieve more lowering of blood glucose (HbA1C <6%) and the control group, called the standard group, used lower insulin and were more relaxed about lowering blood glucose (HbA1C between 7% and 8%). The trial had to be stopped early because the higher insulin group had a higher death rate (first (Total mortality) row in this table: 7.5% vs 6.5%). Notably, risk increased proportionally with increasing insulin dose: (risk (hazard ratio) for heart-related deaths was observed to increase at least 2-3 fold per 1 unit/kg/day increase in insulin dosage). Also, the intensive group had a higher weight gain.
Perhaps if external insulin injections were not invented, or scientists did not jump to conclusions, dietary therapies that reduce the need for insulin may have remained the state-of-the-art approach to diabetes management. Unlike people doing insulin injections, people going on fasting or traditional-foods-based ketogenic diets for diabetes risk management frequently report a reduction in weight, joint pains, headaches, blood pressure, digestion, frequency of cold/flu infections. I have personally experienced ALL of these and so did many people I know personally. Recently, many trials have confirmed many of these modern “crowd wisdoms” and the forgotten wisdoms from the pre-1950 era:
Therapeutic use of intermittent fasting for people with type 2 diabetes as an alternative to insulin
So, compared to the traditional diet-based approach for diabetes management, the universal adoption of insulin-treatment of the disease very likely made life (both quality and quantity) worse for the millions of diabetics around the world who used insulin injections. (That said, newer drugs, like metformin, SGLT2 inhibitors, work in a different way and may genuinely improve outcomes even in the long term.)
What went wrong:
Recall the following fundamental problems with biomarkers:
- just because a biomarker is associated with some adverse outcomes and reducing it by some techniques improves outcomes, doesn’t mean the biomarker causes the problem.
- even when the biomarker is causal, it may not be the root of the problem, in which case, “optimizing” the biomarker may worsen the root of the problem in the long run.
These 2 misunderstandings are at the core of what went wrong in this case:
-
High glucose level was associated with adverse outcomes. Lowering glucose via diet and exercise slowed/stopped/reversed the progression towards those outcomes. These methods lower both glucose and insulin levels. However, many of the bad outcomes, e.g. joint pain and heart disease appear to be caused more by high insulin levels than by high glucose levels. Thus, even if a biomarker (glucose) is associated with bad outcomes and some methods of lowering that biomarker improves outcomes, doesn’t mean that biomarker is the cause of those bad outcomes.
-
High glucose level is still believed to be the cause of many diabetic complications like increased clotting, eye damage, weakened immune system. Even if it is causal for those outcomes, it is likely not the root cause. Attacking it by insulin instead of the root cause (no place to store excess energy) only makes it worse in the long run: it is well known that insulin therapy gradually makes the patient worse (needing more and more insulin dosage to keep the same glucose level followed by tragic complications). This is probably because insulin does not create new fat cells: it forces glucose into existing fat cells thereby eventually damaging them and making them even more resistant to insulin.
Case 2: LDL/HDL in heart disease
LDL cholesterol has a similar decades-long story in the context of heart disease. The above two mistakes also happened in this setting. However, the main goal of this example is to illustrate the third fundamental problem with biomarkers, as mentioned in the beginning: 3) in some cases, there seems to be no good scientific technique for ascertaining whether a biomarker is causal. Another goal is to highlight the damage to human health caused by our blind pursuit to lower cholesterol. For several months of my life, the only question I cared about was: does LDL cause heart disease. After scouring the literature and thinking about it day and night, my unfortunate conclusion is that there is no good technique in the playbook of modern medicine to find an answer. Let us look at the types of evidence presented by those who claim LDL is (not) causal in heart disease –including Randomized Controlled Trials (RCTs) – and why each of them is significantly unreliable.
LDL in coronary artery lesions
LDL particles are almost always found in coronary artery lesions. To many lipidologists, this is a proof that LDL causes heart disease. By this logic, firemen are almost always found at fires thus they cause the fires. It could be that LDL is needed to heal/repair the arteries damaged by something else, e.g. smoking which is much more strongly and consistently correlated with heart disease independently of LDL. There are many experiments suggesting (but not conclusively proving) the latter hypothesis: for example, in this rabbit experiment, researchers damaged the inner lining of the abdominal aorta and studied how it heals. They found:
The most intense radioactivity for both lipoproteins occurred in the region of the leading edge of the endothelial islands where active endothelial regeneration was in progress. The overall distribution of native and MeLDL accumulation was the same. The results suggest that low density lipoproteins are accumulated in areas of active endothelial regeneration by a mechanism that does not involve the high affinity LDL receptor.
Rabbits are not people and abdominal arteries are not coronary arteries (arteries that supply blood to the heart). So this experiment should not be considered a conclusive proof.
Similarly, in this rat experiment, they crushed the rat sciatic nerve and observed the nerve repair:
After a crush injury, as axons died and Schwann cells reabsorbed myelin, resident and monocyte-derived macrophages produced large quantities of apoE distal to the injury site. As axons regenerated in the first week, their tips contained a high concentration of LDL receptors. After axon regeneration, apoE and apoA-I began to accumulate distal to the injury site and macrophages became increasingly cholesterol-loaded. As remyelination began in the second and third weeks after injury, Schwann cells exhausted their cholesterol stores, then displayed increased LDL receptors. Depletion of macrophage cholesterol stores followed over the next several weeks. During this stage of regeneration, apoE and apoA-I were present in the extracellular matrix as components of cholesterol-rich lipoproteins. Our results demonstrate that the regenerating peripheral nerve possesses the components of a cholesterol transfer mechanism, and the sequence of events suggests that this mechanism supplies the cholesterol required for rapid membrane biogenesis during axon regeneration and remyelination.
Again, rats are not people and nerves are not coronary arteries. But these do suggest that serious thought should be given to the possibility that LDL particles are found in coronary artery lesions because they are there to repair the damage caused by something else (e.g. smoking/pollution/high blood glucose).
A fundamental problem with these mechanistic arguments is that humanity has a very incomplete understanding of how the human body works. Thus we will likely make incorrect decisions because of the largely incomplete understanding: e.g. some other unknown mechanisms may overcompensate our actions: e.g. antibiotics overuse causing antibiotics resistance and potentially worse outcomes overall. Also, it is possible that some factors (e.g. high insulin) can make the repair process very slow or dysfunctional and that such prolonged/dysfunctional repair can cause harms in the long run, even though the repair process itself is not the root cause of the problem.
Correlation between LDL-C and heart disease
LDL is correlated to heart disease in many observational studies. In the early days of heart disease research, scientists took this as evidence that LDL causes heart disease. However, the direction of causality could be completely opposite, as explained above.
Also, in older people, cholesterol is either not correlated with mortality (earlier death), or inversely correlated (see Honolulu Heart Program or this broader meta-analysis on LDL and mortality in the elderly). The LDL-hypothesis, which has been the foundation of heart disease research, is so deeply ingrained in medical textbooks and in the minds of most heart disease researchers that they are unable to consider the possibility of LDL being useful. So, instead of investigating the possibility of LDL causing longevity in elderly, they explain the inverse correlation by claiming without evidence that this is because other diseases like cancer cause a lowering in LDL levels.
Most observational studies on cancer survival and heart disease risk factors find that LDL is correlated with better survival in cancer patients, even though all other heart-disease risk factors (e.g. diabetes, high blood pressure, heart failure, stroke, atrial fibrillation) are typically correlated with worse survival. For example, this observational study, which had an impressive 1.8 million person-years of followup, found that high cholesterol (dislipidemia) was associated with 27% better survival. When LDL is correlated with something bad, they always claim that LDL caused them. So, when LDL is correlated with better outcomes, if they were unbiased, they should have seriously considered the possibility that LDL caused the better outcomes, but there they instead claim the opposite causal direction without evidence: that the disease (e.g. cancer) somehow lowers LDL. This is despite mounting evidence that LDL plays an important role as a part of the immune system in fighting viruses and thus may play a role in fighting cancerous growth.
As the above discussion shows, if X is correlated to Y, one cannot conclude the direction of causality (X causes Y or Y causes X). Also, it could be possible that some other factor Z causes both X and Y. Also, if the health authorities have been recommending X (e.g. fearing LDL/saturated fat) for decades, health conscious people (who haven’t dug deep into the medical literature themselves) would do not only X but also other really very healthy things like exercise, smoking cessation. Thus, even if X is slightly harmful, it will likely be correlated with better outcomes. Fundamentally, observational studies cannot judge causality.
Even if the correlation is inconsistent and often in the opposite direction, “scientists”, especially the biased ones continue to hang on to their causality theories.
Randomized Controlled Trials
RCTs (Randomized Controlled Trials) are a great way to judge causality of an intervention, say X. Take a huge group of people, uniformly randomly assign people to either the intervention group or the control group. Do the intervention X on the intervention group. Because the groups were formed uniformly randomly, if the groups are large, the chances of any factor other than X impacting outcomes is negligible. So significant differences between outcomes in the two groups is very likely caused by the intervention X. The problem though is that if the intervention X is composed of many things, it is almost impossible to judge the causality of the individual pieces. This is precisely the problem with the RCTs on cholesterol lowering techniques. As we see below, all known cholesterol-lowering techniques affect our bodies in many ways other than the cholesterol lowering. So, it is impossible to tell whether the benefits or harms in these RCTs were due to cholesterol lowering or something else. Another problem is that the outcomes are very inconsistent and even when benefits are found, they are relatively small and prone to biases.
Diets trials
The most recommended dietary intervention to lower LDL is to replace saturated fats (e.g. butter) with vegetable oils which are rich in omega-6 polyunsaturated fats (PUFAs). This replacement definitely lowers LDL. But it also does many other things. For example, it increases the concentration of omega-6 PUFAs in LDL and many other body cells. Omega-6 PUFAs, with multiple double bonds (poly-unsaturated), are more prone to oxidation. Also, on heating, oils higher in PUFAs produce higher concentrations of polar compounds and oxidative by-products. It has been hypothesized that PUFAs are preferentially converted to ketones, which may be important because ketones are suspected of many longevity benefits. Also, some vegetable oils used in trials, e.g. soybean oil, are rich in vitamin K1, which affects the blood clotting system in a positive way (newborns are given vitamin K1 drops or shots to prevent potentially fatal bleedings). Thus, the substitution of traditional saturated fats (butter/lard) by vegetable oils affect our bodies in many, many ways. Thus, even if RCTs on replacement of saturated fats by vegetable oils consistently showed mortality benefits, we won’t know which effect was responsible and the conclusion we could draw would be that to increase lifespan, one should replace saturated fats by vegetable oils, not that one should reduce LDL concentration.
However, the RCTs are not even consistent. Some of the most rigorous RCTs, e.g. Minnesota Coronary Experiment (MCE) actually had slightly more deaths in the intervention group (butter -> corn oil), despite lowering cholesterol (mean change from baseline −13.8% v −1.0%; P<0.001). The increase in death rate was small and not statistically significant, but a statistically significant decrease was expected: one of the principal investigators told a science journalist that he sat on the results for 16 years and didn’t publish because “we were just so disappointed in the way they turned out.” Also, further analysis showed:
{kind=link}
There was a 22% higher risk of death for each 30 mg/dL (0.78 mmol/L) reduction in serum cholesterol in covariate adjusted Cox regression models (hazard ratio 1.22, 95% confidence interval 1.14 to 1.32; P<0.001). There was no evidence of benefit in the intervention group for coronary atherosclerosis or myocardial infarcts.
About the latter, not all the autopsy files were recovered. However, if the autopsy examination showed less heart disease (e.g. number and volume of lesions), the investigators would have been shouting it from the rooftops, instead of hiding the data for decades.
I said MCE is one of the most rigorous diet RCTs, because there was great control/tracking of what the subjects ate, because it was done in a mental hospital. Also, replacing butter by corn oil was the only intervention. Also, outcomes were judged in an double-blind design. In contrast, many other trials often cited to claim that saturated fats should be replaced by vegetable oils did several more interventions concurrently. For example, the interventions in the STARS and the OSLO diet-heart trials also included adding fruits and vegetables. The latter also included reducing grains and sugar, drastically reducing trans fats (which are now banned in the US), adding fish, cod-liver oil, which is a great source of vitamins A/D which are associated with better overall health outcomes. I was surprised (and saddened about the quality of science in the medical area) to notice that the main paper of the OSLO study does not even mention this in the methods section. Apparently, only the supplementary material, which I could not get a hold of, mentions these extra interventions.
On the flip side is the LA veterans RCT, which is the other double blind RCT. The intervention was not as clean as the Minnesota experiment, but also not as confounding as the OSLO trial: for example, there the intervention group’s diet had double the iodine content, and a higher Vitamin E (α-tocopherol) content. However, a great aspect of this trial is that it ran much longer: 8 years. There were fewer heart-related deaths, but more cancer-related deaths in the intervention group. The overall death rate was the same. Even though there were fewer heart-related deaths in the experiment group, autopsies found:
Gross grading of the extent of atheromata in individuals who died and were autopsied failed to reveal significant differences between the two groups of subjects. The same was true of arterial total lipid and calcium concentrations. Relative abundance of major lipid fractions in coronary atheromata and circle of Willis appeared to be independent of diet.
The dietary guidelines designed to lower cholesterol very likely unleashed an epidemic of many diseases that were unknown just a few decades ago, e.g. the non-alcoholic fatty liver disease (NAFLD), even though alcoholic fatty liver disease, which has the same clinical manifestation (e.g. elevated AST/ALT, fat in liver on ultrasound) was well known for several decades; thus it is hard to say that the NAFLD epidemic is solely because we recently became better at diagnosis. For example, 22.4% of the children aged 5-10 in a North Indian school had ultrasound-confirmed fatty liver disease. I myself had ultrasound-confirmed fatty-liver disease in my early 20s (my ALT was often over 100). I and many others I know were able to later reverse it by doing the exact opposite of the guideline: by replacing carbs and omega-6 vegetable oils by saturated fats. In the previous year, my ALT has always been below 20. Indeed, animal experiments suggest that as long as linoleic acid (the primary omega-6 fat in vegetable oils) is kept very, very low, even large amounts of alcohol cannot damage liver. I suspect substituting sugar by alcohol in that experiment will produce the same result, and sugar is something Indian kids do consume a lot. (Linoleic acid should not be confused with α-Linoleic acid or conjugated linoleic acid: those are different compounds and have different, often opposite effects: even peer-reviewed and published medical papers confuse them).
Also in the fear of cholesterol, foods like egg yolks and liver, which are extremely rich in vitamins (A/D/E/K2/B12/Choline/folate) were demonized due to their cholesterol content, even though it was known decades ago that dietary cholesterol does not even affect blood cholesterol. These have likely played a role in our mental-health epidemic and in several other neurological issues. My recent Uber driver was from Mexico. I steered our conversation towards traditional pregnancy practices in Mexico. He said that traditionally, after birth, women were given liver to combat depression. Choline from liver and egg yolks also can cure fatty liver. For example, previously, kids on TPN (vein-injected nutrition because of digestive problems) used to suffer liver (hepatic) damage, which was cured perfectly just by adding choline to the TPN formula.
In summary, even simple dietary changes like replacing butter with corn oil or soybean oil affects our bodies in many, many ways other than changing our LDL concentration. So the results of such trials, even if consistent, cannot tell whether the effects were due to the changes in LDL concentration or something else. On top of that, many dietary trials do more changes than just fat substitutions, further confounding things. Thus, none of these give us reliable evidence on the causality of LDL concentrations. Also, many important nutrients, e.g. choline, B12, folate, suffered in the crossfire resulting in disastrous health consequences.
LDL-lowering drug trials
Several drugs have been used to lower LDL. Several large trials have been conducted to assess the mortality benefit of such drug therapies. Drug trials suffer from the same problem as the diet trials above: all drugs (as far as I know) affect our bodies in many ways. For example, statins, which are the most widely promoted LDL-lowering drugs, not only lowers LDL, but also lowers hs-CRP, a marker of inflammation, which is independently associated with heart disease, often more strongly than LDL, e.g. in this small study of Indians. Even in the US, in the Jupiter trial, the benefits correlated better with the reduction in hsCRP, rather than LDL. Also, statins reduce triglycerides (at least when >250mg/dl) and increase HDL. In this large meta-study of 32 studies of heart disease incidence (total > 0.5 million person-years) in the Asia Pacific region, higher triglycerides and lower HDL were a much better predictor of heart disease death than LDL. Statins also reduce ferritin, which is a biomarker of metabolic syndrome; in many trials of peripheral artery disease, the positive outcomes correlated more with the reduction in ferritin than the changes in LDL. Blood donation, which reduces ferritin, is correlated with reduction risks of heart attacks by 86%, which is a much greater risk reduction than statins (<50%). (Unfortunately, because this was not an RCT, there may be confounding factors. I could not find an RCT evaluating blood donation, which is incredibly safe/cheap/simple, in reduction of heart attack risk, despite the fact that billions of dollars have been wasted in studying so many drugs that turned out to be harmful.)
So, even if statins consistently reduced mortality, we cannot say the benefits are due to the lowering of LDL, or that of reducing hsCRP or ferritin, or triglycerides, or increasing HDL. Unfortunately, statins, the best drug (w.r.t. evidence of mortality benefit) cardiologists have to offer do not consistently improve meaningful and objective outcomes such as lifespan, as we will see below. Also, many (most?) drugs investigated to reduce LDL never produced meaningful and objective outcomes such as lifespan, and often only did harms. The negative trials have almost been forgotten by most in the cardiovascular research community. Those who remember them, some of whom have struck me as the most logical and knowledgeable voices, are often labeled quacks and ostracised.
Clofibrate was one of the drugs studied in a large RCT (n>10,000). There was a higher death rate in the intervention group, even though it reduced LDL by 9% (which is modest compared to modern drugs). In the ILLUMINATE trial, a CETP inhibitor reduced LDL by 25% and increased HDL (“good cholesterol”) by 75% increased deaths so obviously that they decided to stop the trial mid way. Just like all other drugs this CETP inhibitor affects human bodies in many, many ways. So, believers of the LDL causes heart disease were quick to claim that the higher death rate was due to some factor other than LDL-lowering. I think they were probably right. My problem though, is that when an LDL lowering drug reduces mortality even a tiny bit, they jump on it confidently as proof that LDL causes heart disease. In a sense, LDL has become God: when something good happens, people always credit God for it. When something bad happens, they never blame God for it. The ILLUMINATE trial should have taught scientists the limitations of relying on biomarkers, but apparently they haven’t learned the lessons.
More recently, very powerful LDL-lowering drugs have been invented. For example, Repatha can lower LDL down to <30mg/dl, lower by at least 3 times than the earliest recorded median LDL levels, which itself was probably lower than the levels several centuries ago because by the time cholesterol measurement started, people had already switched from traditional saturated fats to vegetable oils to some extent, which reduces LDL. Many have argued that there was less chronic disease (diabetes, heart disease) 1-2 centuries ago, but I don’t have good evidence to back it up. To me, my generation does appear to get the diseases of age much earlier than my parents’ or grand-parents’ generation. Regardless of the history, I hope everyone agrees that <30mg/dl is an incredibly low level and is unprecedented in human history and even in the history of other species of even herbivore mammals. It is probably at the lower limit of biological safety. Yet, in the Fourier trial, there were more deaths (see the row death from any cause in this table) in the Repatha intervention group (444 vs 426, each group had about 13,780 subjects). The increase may be due to chance, but we should have seen a significant decrease if LDL causes heart disease.
{kind=link}
However, there were 27% fewer heart attacks (myocardial infarction, 468 vs 639) and 22% fewer stenting/bypass surgeries needed in the Repatha group (750 vs 965). On the surface, this looks like a small but very useful benefit. However, it is important to note that the first thing they check in a heart hospital is your LDL level, which will immediately unblind the group (intervention vs control) of the patient. Also, whether a primary care doctor suspects that some chest pain is heart related is likely to be biased based on the patient’s LDL level. Many angina (heart-related chest pain) episodes do go away on their own; in a person with low LDL, their primary care doctor may not even suspect heart disease. Also, myocardial infarctions can often be asymptomatic or unrecognized. So, there is a possibility of bias, especially that these trials were done by the drug manufacturer and academics on their payroll. In contrast, any-cause-death is very objective, and perhaps the thing most people care about: it is not great to avoid heart disease and get cancer or some neurological disease. Also, several trials, such as COURAGE, ORBITA, have shown that most of the stent procedures (e.g. in those not having an active heart attack) are unnecessary anyway. So, the number of stent procedures is an unreliable metric.
In my opinion, the fact that such a spectacular LDL lowering did not improve death rate and only had small improvements in subjective outcomes prone to bias, is a big red flag in the LDL causality hypothesis of heart disease. However, it is possible, although very unlikely, that Repatha helps massively by massively lowering LDL, but also does some other thing that is equally or even more massively harmful to health, resulting in the two effects almost cancelling out. Thus I cannot argue that the Fourier trial conclusively proves that LDL does NOT cause heart disease.
In many studies, statins, which are not as effective as Repatha in lowering LDL, have actually shown reduction in death rate. The most spectacular improvement was seen in the 4S study, where there was a 30% reduction in death rate using simvastatin. In the JUPITER trial, the death rate was reduced by about 20%, but it selected patients with high hsCRP, regardless of the LDL level. There are many other trials that observed reductions in death rate, but those reductions were smaller than 20%. However, in many other trials no benefit was seen in the death rate: In the TNT trial, 10000 subjects were randomized into either a 80mg Lipitor (a popular statin, aka Atorvastatin) group or a 10mg Lipitor. The 80mg Lipitor group had a 34% lower LDL than the 10mg group. Yet, the death rate was the same: 284 in the 80mg group vs 282 in the 10 mg group.
{kind=link}
In the STABLE study, they randomized 312 patients 2:1 into either a 40mg Crestor (Rosuvastatin) or a 10mg Crestor group. They used intravascular ultrasound to image the lesions in the hearts. In this method, they insert a catheter with a tiny ultrasound tip into the wrist artery and guide it up towards the arteries in the heart. Using the ultrasound tip, they can look into the heart’s arteries: extremely impressive technology, I must admit. At 12 months, the 40mg group had a greater reduction in LDL-C (59 vs 80). In both groups, there was a very slight overall reduction (regression) in the total volumes of artery lesions. However, there was no difference in the reductions between the 40mg group vs the 10mg group. Thus, just like in the Jupiter trial, the improvements correlate better with the reduction in hsCRP, which was reduced equally in both groups, instead of LDL. To me, this suggests that the tiny beneficial effects of statins, if any, are likely due to some mechanism that reduces hsCRP (not necessarily directly due to hsCRP reduction), rather than the LDL reduction.
Similarly, in this RCT, patients were randomized into either pitavastatin or atorvastatin. As expected during the 240 week followup, the cholesterol levels were similarly reduced in both groups and the resultant LDC-C levels were similar in both groups. However the pitavastatin group had a 2-3x lower rate of the composite of cardiovascular death, sudden death of unknown origin, nonfatal myocardial infarction, nonfatal stroke, transient ischemic attack, or heart failure requiring hospitalization.
In ALLHAT-LLT (n = 10,355; patients with high cholesterol and high blood pressure) , AURORA (n=2,776; severe diabetics on hemodialysis), GISS-HF (n=4574, patients with heart failure) the effect of statins not just objective endpoints like death rate, but even subjective endpoints such as the rate of myocardial infarction, stent/bypass surgeries had P>0.05 (aka not statistically significant).
In summary, if we average over all statin trials, we will indeed likely see a tiny benefit in death rate. But, as described above, statins affect us in many ways that are also correlated with lower heart disease, often more strongly correlated. Thus, the tiny benefit cannot be attributed to LDL-lowering. Also, in randomized trials of low vs high dose statins, which are least likely to suffer from confounding factors or reverse causality, despite huge differences in LDL-lowering, differences in death rate or reduction in volumes of lesions (using intravascular ultrasound) were the same, suggesting the tiny benefit is likely due to some mechanism(s) unrelated to LDL lowering.
Given that most of the LDL-lowering trials were done by industry and academics on their payroll, there is a chance of bias and fraud in some of the trials. Most medical guidelines use the “weighted averages” of trial results to make decisions for the public. Even a few fraudulent trials can significantly influence the average. Perhaps using the median would be a better idea? There is no dearth of confirmed fraud in industry trials, especially withholding negative data on drugs: Vioxx scandal, Tamiflu scandal, EXCEL scandal. There is evidence of fraud even in the statin trials. For example, the rate of muscle pain is 1% in trials sponsored by the statin drug manufacturers, but 10% in other trials. (I personally experienced muscle pain even while taking the lowest dose statins. Three times, my pain would start within a day of starting statins and stop within 2 days of stopping them.) Most doctors I have talked to, many who themselves recommended statins to me, estimated the muscle pain rate in their patients to be over 20%. Boston Heart Diagnostics claimed that the rate is 29% when selling a genetic test to identify those who will develop muscle adverse effects with statins.
Just like the dietary approaches to reduce LDL, many drug approaches to lower LDL have very likely damaged the health of many people worldwide. The side effects of statins are not limited to muscle pains, which were reversible in my case. They can also damage the heart. In this trial, doctors had the unbiased critical thinking skills to try temporarily stopping statins in 142 patients with heart failure with no known cause. The heart failure of most of them actually improved on stopping statins and supplementing CoQ10, which statins deplete in muscles (heart is also a muscle). So did memory loss, which many people claim to experience on statins. Compared to other drugs, some statins users were 100+ times more likely to report ALS in the FDA Adverse Event reporting system, which is a very rare but very devastating disease. Cancer is correlated with lower LDL. The LA Veterans study where omega-6 PUFAs were used to lower LDL, saw increased rates of cancer, but only after 6 years. In an observational study, LDL-C values were lower in cancer subjects than matched controls at each point of assessment throughout an average of 18.7 years prior to diagnosis. Most trials of LDL-lowering drugs did not last long (many large ones were stopped as early as 2 years). We won’t find what we are not even looking for. Also, those in charge of looking for it have billions to lose if they find it. So, even though I am not aware of any drug trial reporting statistically significant increases in new cancer diagnosis, the possibility should be seriously considered.
{kind=link}
The best longevity interventions increase LDL
Among all the interventions known to improve longevity in a wide range of species, nothing has more evidence than periodic fasting (often known in medical literature as calorie restriction). Also, there are several anecdotes (e.g. here) and even a book documenting how in many patients, a prolonged (several-days) fast cured angina (chest pain on exercise typically caused by insufficient supply of blood to heart because of narrowed arteries). Prolonged fasting drastically increases LDL, at least for a few weeks even after the fast. For example, a 7-day fast significantly increased LDL by 66%. Also, this increase was correlated with the weight loss and is probably caused by upregulation of the fat-burning pathway. Over several weeks, the LDL increase subsides. However, many of the disease reversals by prolonged fasting – including revesal of angina – happen during or within a week of the fast, at which time, the LDL is dramatically elevated. For example, I continuously had skin sores at the back of my neck for 7-8 years, which completely disappeared by 3-5 days after my 5-day water+salt fast during March 2018. Those sores have not come back yet. Fasting dramatically lowers both insulin and glucose, which may be a reason why it is such a magical healer.
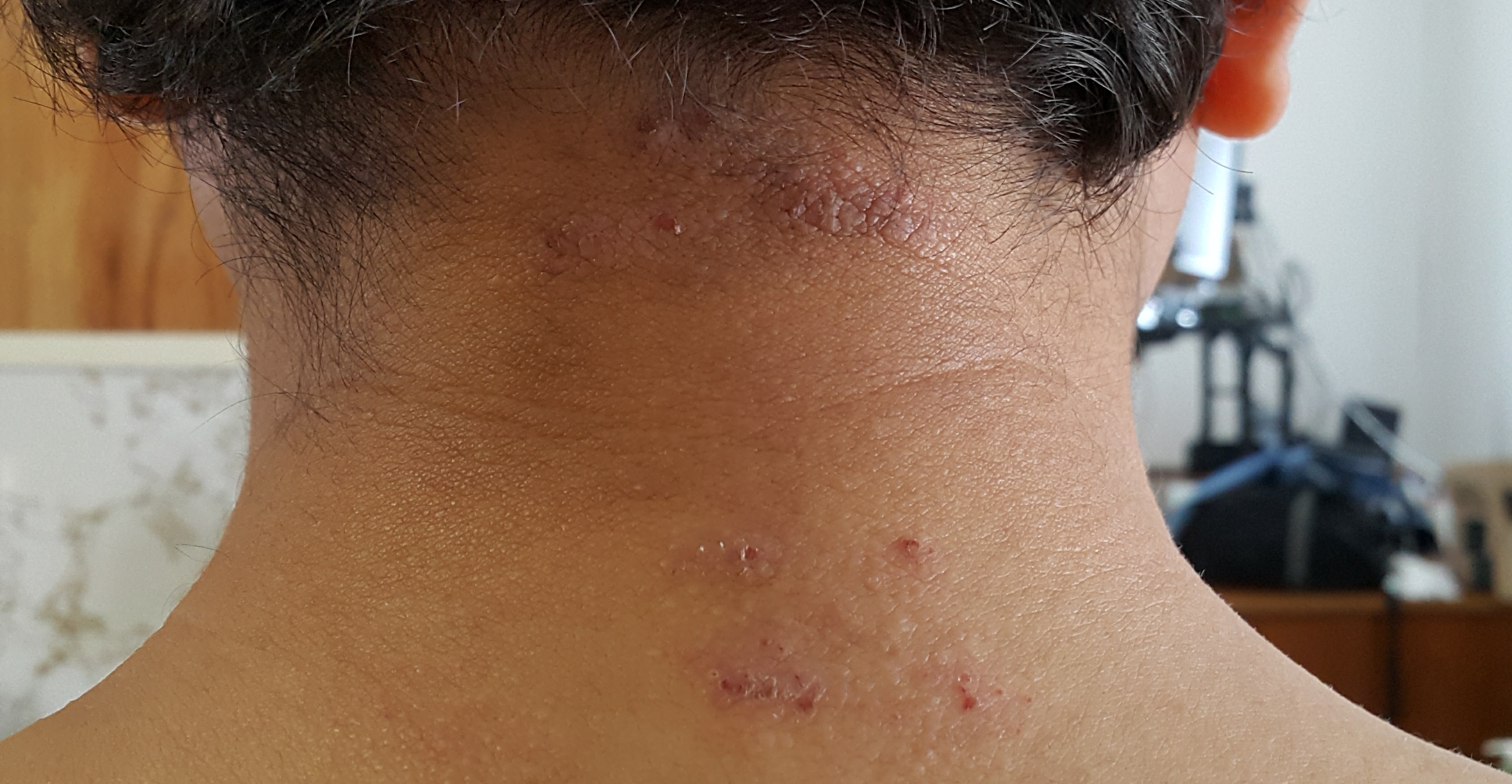{kind=link}
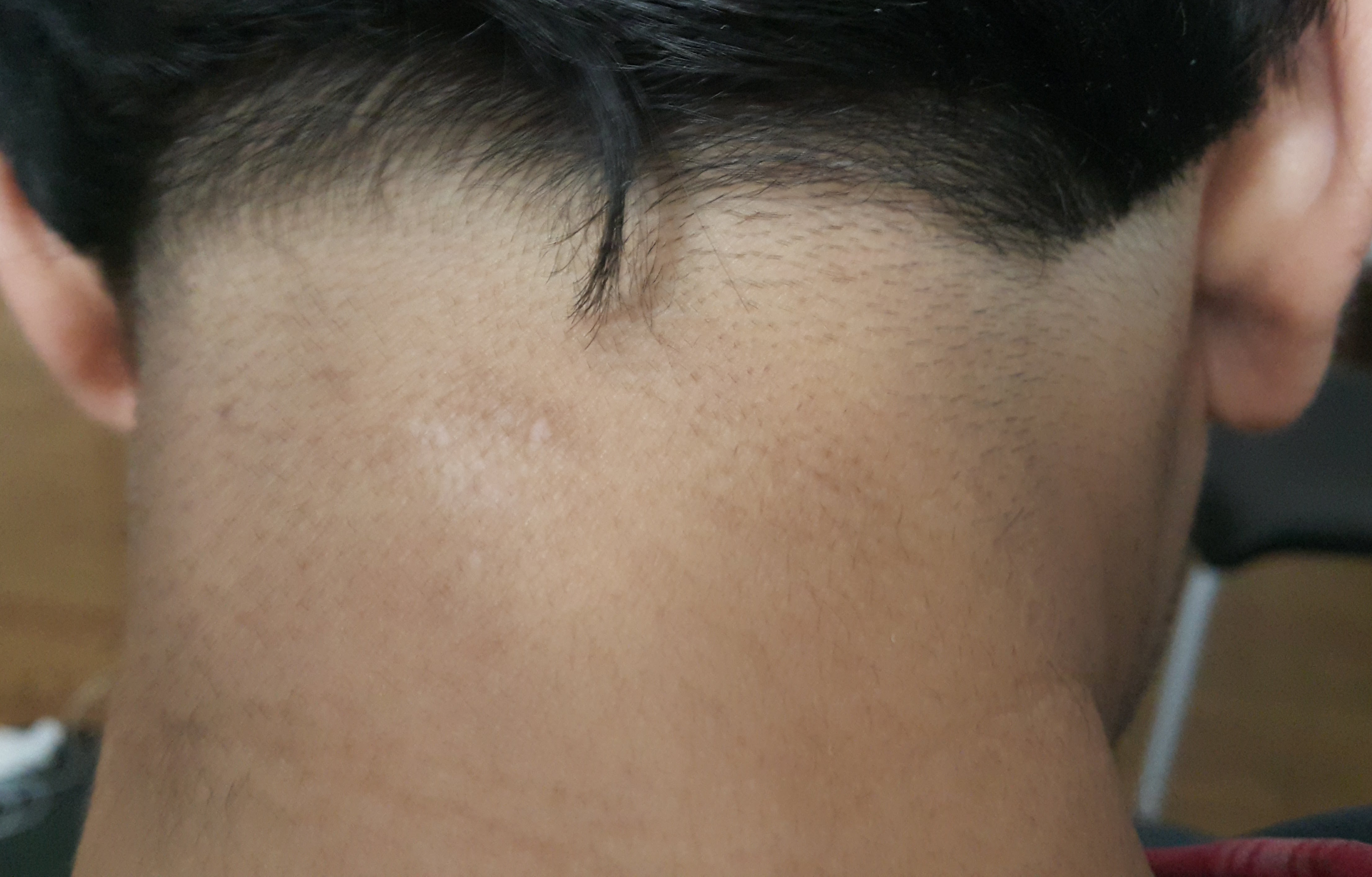{kind=link}
Many longevity experts consider Rapamycin (aka Sirolimus) to be the most promising longetivity drug, with mechanisms similar to prolonged fasting (e.g. autophagy). In various species, it not only improves lifespan, but also improves various aspects of heart disease. In this randomized trial of human patients receiving heart transplants, at 6 weeks, 6 months, and 2 years, they observed the inside of heart’s arteries by intravascular ultrasound (guiding a catheter with ultrasound tip from wrist’s arteries to heart’s arteries). From 6 weeks to 6 months, they actually observed a tiny regression of lesions (plaque burden) in the Sirolimus group, even though the plaque burden increased significantly in the other group. This was despite the fact the patients’ LDL significantly increased after the transplant, especially till month 6. Indeed, this is not an isolated finding, and it seems well known that Sirolimus (mtor inhibitors in general) increase LDL despite improving heart disease.
{kind=link}
Fasting and Rapamycin affect our bodies in many, many, ways other than increasing LDL. It is possible that some other beneficial mechanism outweighs the increase in LDL. So, these cannot disprove the LDL causality. However, statins also affect our bodies in many, many ways, as explained above, so the tiny benefit of statins (which does not correlate with LDL-lowering on dose randomization) should also not be taken as proof of LDL’s causality in diseases.
In practice, none of this matters much anyway. Ultimately, what humans do are specific interventions (e.g. fasting, take drug X, replace food U by V) and long-term RCTs can reliably judge the causality of those interventions in improving outcomes that matter to people (e.g. lifespan or non-miserable lifespan). Biomarkers should just be used to postulate potentially new interventions to investigate in clinical trials. Biomarkers themselves should not become the end goals, as LDL has become. I hope people understand that dying early and in pain, but with biomarkers that perfectly match the biases of “scientists” is not better than living a longer and more fulfilling life with biomarkers that don’t match so perfectly.
Main Takeaway:
If your doctor gives you a medicine X to increase or decrease the levels of Y so that the level becomes less correlated with a disease D, ask them for direct evidence (ideally an RCT) that X reduces mortality or some other outcome you care about, e.g. reduced suffering from pain, coupled with evidence that the medicine does not increase mortality at least during a few years. You definitely don’t care directly about the blood levels of various compounds in your body.
Found something wrong in this page? Please make an issue here or submit a pull request to this file.
Acknowledgements:
This article has benefited from ideas in talks/tweets/articles of Ted Naiman, Nadir Ali, Chris Masterjohn, Gabor Erdosi, Ivor Cummins, Tucker Goodrich, Malcolm Kendrick, and perhaps many other doctors/scientists/citizen-scientists who are passionate about this topic. However, I dont agree with them on everything and I have verified their claims by independently reviewing the published literature. Mistakes in this blog are all mine.
Disclaimer: This article is for informational purposes and does not constitute medical advice.